热线:021-56056830,66110819
手机:13564362870
热线:021-56056830,66110819
手机:13564362870
2.1.4热处理对催化性能影响
高温热处理是改善大环化合物氧还原催化剂的催化活性和稳定性的有效途径。LEFVRE等认为,最佳的催化活性发生在500——700℃,催化活性位是MN4/C;另外一个活性位为“高温活性位”,温度大于800℃。FAUBERT等分别将FeTPP和CoTPP吸附在碳黑上,在Ar下进行热处理。当温度在500——700℃时,活性位为MN4或是一些分子碎片中包含了部分N与金属键合物,其稳定性较差;当温度大于900℃时,具有更好的活性和稳定性,且N与金属的键联消失,大量的金属颗粒被石墨包围。ALVES等发现CoPc/C在850℃热处理后催化活性最高,此时存在直径为2 nm的Co簇。LALANDE等把酞菁铁和四羧酸酞菁铁负载到碳黑上,并在氩气下100——1100℃热处理,发现900℃以上的热解产物具有高催化活性和稳定性。GOJKOVIC等对FeTMPP-Cl/BP研究表明:在200——400℃范围内开始分解;当T>400℃时,Fe(Ⅲ)/Fe(Ⅱ)氧化还原峰电对消失;当T>700℃时,有金属Fe粒子形成,此时循环伏安曲线上存在一个氧化峰,这是热处理形成的金属Fe的溶解过程。热处理温度为600——800℃时,钴卟啉具有最好的活性。酸性中,Co-C-N在临界热处理温度725℃处开始表现有催化活性,此时催化剂转变成β-Co和含N石墨碳的异相结构;碱性中,Co-C-N在500℃就开始表现有催化活性,在850℃处活性最佳。
2.1.5催化活性位探讨
催化活性位的确认一直是过渡金属大环化合物催化剂研究中倍受关注的课题。关于高温热解条件下的催化活性位存在很多争议:(1)高温热解后的催化活性位仍然是FeN4结构,如图2所示;(2)高温热处理后残留的N与吸附在C表面上的过渡金属离子相互作用形成C-Nx-M物种,但FAUBERT认为Fe离子与C-Nx基团相结合对ORR的催化活性无影响;(3)N原子和过渡金属原子共同构成了催化活性位,而单纯由含N或过渡金属前驱体制备的物质无催化活性;(4)热处理后形成具有化学表面基团的高活性C,并认为金属的存在只是催化了这种高活性C的形成,金属可能是起到在高温裂解含N和C前驱体时催化形成活性位的物质,更多的石墨边缘位和吡啶氮有利于提高催化活性,Fe颗粒的形成可能有利于边缘位C纳米结构的生长,如图3所示;(5)在高温热解下形成的石墨包覆金属颗粒具有催化活性,石墨层可保护金属颗粒免于外界环境的腐蚀。
2.2过渡金属——氮/碳类化合物
研究表明,在高温热裂解下不使用大环化合物作前驱物也可以得到对氧有活性的催化剂。GUPTA等首次报道了采用聚丙烯氰(PAN)、金属Co或铁盐和C载体混合后在惰性气氛下高温热处理制备氧还原催化剂的研究。该方法与常规的以含N4结构的大环化合物为前躯体的催化剂制备方法相比,不仅制备工艺简单,而且清洁无污染,因而引起了国内外学者的广泛兴趣。YE等报道了通过碳化含有铁盐或钴盐的多孔PAN来制备Fe或Co气凝胶纳米复合物催化剂的方法。BRON等报道了在Ar或NH3氛围中热处理碳载铁邻二氮杂菲化合物制备氧还原催化剂的研究。WEI等通过裂解高比表面C、乙腈和硫酸钴的混合物来制备催化剂,结果表明,催化活性与N和Co的含量无关,但采用多步法可以得到性能较好的催化剂。OKADA等报道了N、N′——二——8-喹啉三甲基二胺钴和N、N′——二——8-喹啉苯基二胺钴的催化性能随着热处理温度的升高而增加。ALVES等以聚甲基乙烯基酮(PVK)、聚丙烯酸(PAA)和聚苯乙烯(PE)为配体考察了配位作用对催化剂性能的影响。BOUWKAMP-WIJNOLTZ等提出了用乙酸钴、碳黑和各种含N前驱体制备氧还原催化剂的方法。EXAFS表明,这类催化剂有活性位CoN4,但同时也有金属Co的存在。JIANG等研究发现ORR反应路径与配体的结构有关:2、4、6-三甲基苯基配体是2e反应路径,2、6-二异丙基苯基配体是4e反应路径。
另外,SIRK等基于乙二胺和1、2-苯乙二胺合成了两种Co基氧还原催化剂。YUASA等用电化学沉积法在C颗粒表面修饰一层聚吡咯膜(PPY/C),并在其表面吸附Co离子,700℃热处理后获得了在酸性条件下稳定的Co-PPY/C.2006年,BASHYAM等在《Nature》上报道了用化学还原法合成了Co-PPY-C作为PEMFC阴极催化剂的工作,如图4所示。
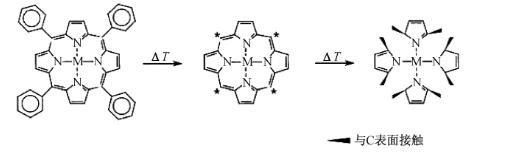
图2 MN4催化活性位示意图

图3不同类型N在C载体表面的示意图
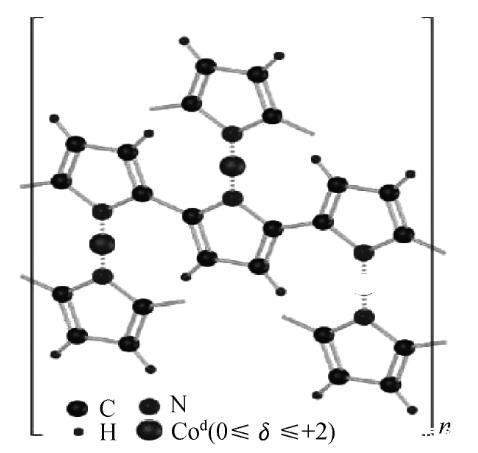
图4 Co-PPY-C催化剂的结构示意图
 相关新闻
相关新闻