热线:021-56056830,66110819
手机:13564362870
热线:021-56056830,66110819
手机:13564362870
导读
开发一氧化二氮(N2O,又作笑气)重新利用的催化化学工艺,对减轻其排放所带来的环境威胁有极大的意义。传统上,N2O被认为是一种惰性分子,由于其活化需要的苛刻条件(>150 oC,50-200 bar),有机化学家难以将其作为氧化剂或氧原子转移试剂。近期,德国马克斯-普朗克煤炭研究所的Josep Cornella教授课题组报道了在温和条件下(室温,1.5-2 bar N2O)将N2O插入Ni-C键,从而提供高附加值的酚类化合物并释放无污染的N2。
正文
温室气体的排放给全球环境带来了极大的威胁,因此温室气体的减排与转化已经成为科学家们研究的热点。从可持续性发展的角度来看,非常需要开发超越传统降解的化学工艺,并将这些气态副产物重新利用,以生产高附加值的化学原料。尽管通过催化策略将CO2或CH4作为有机合成碳源已受到广泛关注,但对导致全球变暖的另一个主要贡献者——N2O,相关研究却少得多。目前仅有几个例子仍然依赖于传统的金属-氧化反应,这需要高温高压或长反应时间(Fig.1a)。上世纪八十年代,Vaughan等人报道了一项开创性研究工作:N2O中的O原子可插入配合物1的Hf-Ph键(J.Am.Chem.Soc.,1987,109,5538–5539),并通过释放N2形成所需的Hf-O-Ph(2),但是O原子插入会出现区域选择性问题,即插入到金属氢化物上形成Hf-O-H配合物(3)。近期,德国马克斯-普朗克煤炭研究所的Josep Cornella教授课题组通过有机金属Ni-介导的Baeyer-Villiger(OMBV)型反应来构建C(sp2)-O键(Fig.1b,right),即N2O与金属中心配位并且亲电O原子插入Ni-C键形成Ni-O-C键,同时释放出N2。而传统的过渡金属催化芳基卤化物合成酚类是通过还原消除形成C(sp2)-O键(Fig.1b,left),并且往往需要在碱条件下进行,存在一定的局限性。在本文中,作者利用有机金属Ni配合物活化N2O的策略(Fig.1c),在温和条件下成功地将一系列芳基卤化物高选择性地转化为高价值的酚类化合物。
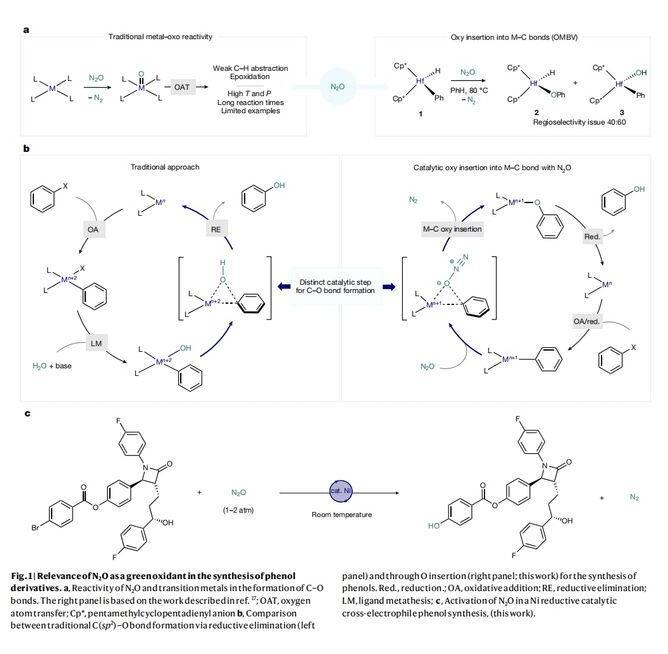
(图片来源:Nature)
根据前人的研究成果:N2O可以与某些膦-Ni(II)配合物反应(Organometallics1995,14,456-460;J.Am.Chem.Soc.1993,115,2075–2077.)。作者合成了氧化加成产物4,并研究了其与N2O的反应性(Fig.2a)。结果表明:在氩气氛围下溶解于DMA中,4发生分解主要生成自偶联产物5以及痕量的脱卤产物6(path a),在Zn等还原剂的存在下,会加速4的分解(path b)。将氩气换成N2O时,4的溶液的亮红色仍然存在,表明分解速率变慢。经过酸处理后,酚7的产率为15%(path c)。在此基础之上,向反应体系里添加Zn做还原剂,可提高7的产率,尤其是添加Zn和NaI的组合时,7的产率可提高至73%(path d)。随后作者对配体进行了筛选发现联吡啶2-位取代的三齿氮化配体对催化活性至关重要,其中三联吡啶(L18)和6-吡唑基-2,2'-联吡啶(L50)效果最好(Fig.2b)。用C-H或S取代N原子会抑制催化活性(L48和L61);吡唑单元的N旁边的空间位阻也会抑制催化活性(L55和L58);吡唑上的缺电子取代基会显著降低酚的产率。为了证明反应中的Ni-ligand的参与,作者制备了配合物10并对其进行了结构表征,使用10作为催化剂,得到酚9(75%),因此证实预配合物具有催化能力。然而,尽管邻位中存在两个Me基团,但在Zn和NaI存在下,反应后用酸性处理后以49%的产率提供了所需的mesitol(12)。为了进一步证明Ni(I)-C(sp2)参与反应,作者将(tBu-terpy)Ni(I)-I(13)与Ph2Zn在N2O下进行了反应,观察到20%的酚(14)的生成。这些发现表明,Ni(II)物种被还原为Ni(I),以及碘盐对C(sp2)-O的形成键非常重要。
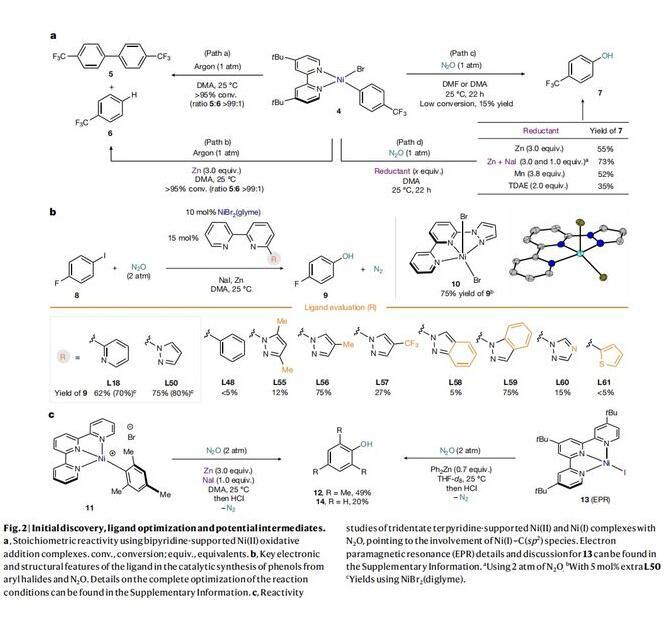
(图片来源:Nature)
在最优反应条件下,作者初步探索了芳基卤化物的底物范围。如Fig.3所示,在对位(9,15,16)和间位(17-19)位置均带有其他卤素的芳基碘化物均可以优异的产率获得相应的酚。CF3(7)、酮(20)、酯(21,24)或腈(22-23)等吸电子基团的存在对C-O键的形成没有任何困难。供电子取代基,如烷基(25)、芳基(26),甚至甲氧基(27,32)和硫甲基(28),也能以良好的收率得到相应的酚。此外,具有苄基C-H键的芴衍生物(29)也适用于酚的合成,但产率有所降低。事实上,茚满酮(31)和1-氯-2-碘苯(30)衍生物产率略有下降,或许是因为邻位的空间位阻会抑制反应性。在该体系中,TIPSO保护的苄醇(33)和二乙基膦酸酯(34)也是可以兼容的。杂环如吲哚(35)、喹啉(36)、咔唑(37)和二苯并噻吩(38)同样可以兼容。另一方面,N,N-二苯基取代的底物形成C-O键后易快速氧化,因此可进一步原位官能团化生成特戊酰基衍生物(39),而具有生物活性的氯贝特衍生物(40)和含有频那醇硼酸酯的底物(41)则得到相应的酚类产物。需要指出的是,底物(28)的反应中还观察到7%的亚砜以及低产率的芴醇(29),这些结果表明氧插入步骤与OMBV-类型反应中的oxo/oxyl-途径相一致。另外,7、9、18、25和34的反应结束后还检测到N2的存在(Fig.3)。当溶剂上的氧用18O标记([18O]DMF,25%18O)时,9中没有观察到18O。另一方面,当使用N15N18O(约23%18O)时,22%±1的O在9中被标记,进一步证明了产物中的O来源于N2O。
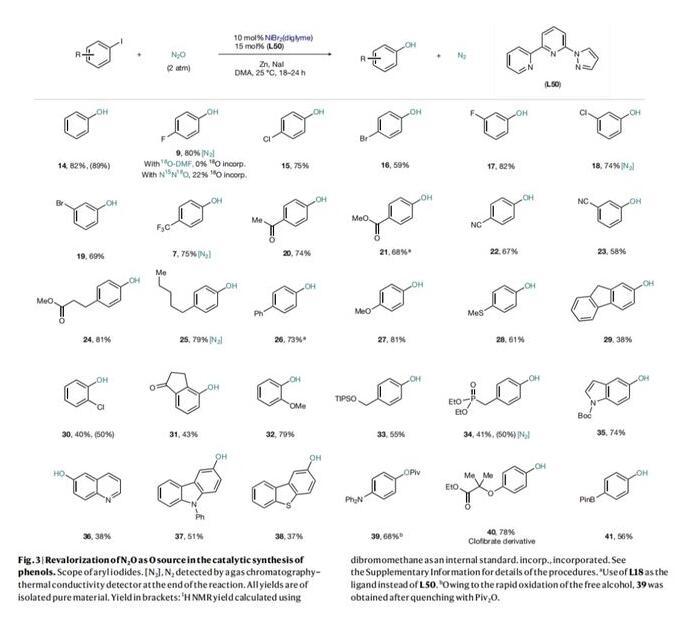
(图片来源:Nature)
在相同反应条件下,更容易获得的芳基溴化物同样可以发生该反应,然而,需要吸电子取代基才能使C(sp2)-Br裂解。如Fig.4a所示,不同的基团如CF3(7)、Ac(20)、CO2Me(21)、CN(22)取代的溴苯、药用相关的酞类(42)、共轭体系(如萘甲酸酯(43)和肉桂酸酯(44)以及芳基甲基砜(45)溴化物均能顺利转化,以中等至较好的收率获得相应的酚类化合物。与目前的光介导过程相比,没有观察到44中双键的异构化,但杂环溴化物与当前方法不能兼容。随后考察含有敏感官能团的复杂芳基卤化物。例如,一种恩格列净衍生物(46)(含有大量容易产生HAT的弱C-H键),一种天然产物丁香酚的衍生物(47),甚至将其放大至5 mmol,产率仅略有降低(66%)。含有饱和N-杂环的底物如哌嗪酮(48)、氮杂环丁烷(49)、吡咯烷酮(阿尼西坦中间体)(50)、去甲托品酮衍生物(51)、胆固醇吸收抑制剂依折麦布衍生物(53)以及帕罗西汀衍生物(54)均具有良好的兼容性。吸电子基团可以促进芳基溴化物的转化,从而实现区域选择性控制(52,78%)。最后,作者将温室气体N2O和CO2用于药物合成,并成功地完成了肌肉松弛剂美他沙酮的形式合成(59,Fig.4c),其中66%的氧源自废气原料。值得一提的是,该方法对于抗乳腺癌和胰腺癌的候选药物巴多昔芬(68)的合成同样适用,即从三种不同的母体卤代物出发,以良好的收率获得三种酚类中间体(64-66),随后65和66经Fischer吲哚合成法得到吲哚中间体(67),后者与64进行反应便可合成巴多昔芬(68),并且68中的所有O原子均来源于N2O。
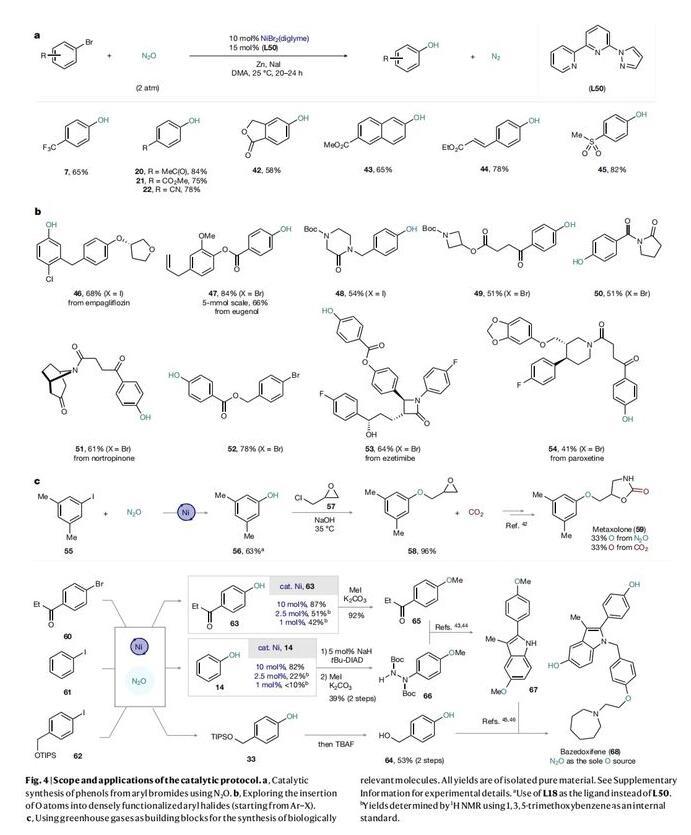
(图片来源:Nature)
总结
Josep Cornella教授课题组将N2O作为绿色氧源,利用有机金属Ni配合物活化N2O,在温和条件下成功地将一系列芳基卤化物高选择性地转化为高价值的酚类化合物。这种独特的有机金属C-O键形成不同于当前基于还原消除的策略,并为芳基卤化物转化为酚类化合物提供了新方法和新借鉴。
