热线:021-56056830,66110819
手机:13564362870
热线:021-56056830,66110819
手机:13564362870
可溶性锰能够沉积在水处理设备的管道、阀门和过滤器上,导致设备的堵塞和性能下降。锰暴露会影响人的神经系统功能,损害人体健康。MnO2作为一种重要的吸附剂,具有十分优秀的表面特性,如高表面负电荷、大表面积和低零点电荷等,且表面含有大量的—OH等供电子官能团,在处理含重金属水等领域受到广泛关注。有研究发现,传统高锰酸钾氧化除锰过程中,生成的原位MnO2胶体对锰的吸附效果比商品MnO2更显著,原位生成的MnO2胶体颗粒表面羟基以及活性吸附位点比商品MnO2更丰富,吸附能力更强。但关于原位生成的MnO2胶体对Mn2+离子的吸附机理以及影响因素研究相对较少。
本研究通过Mn2+与KMnO4溶液反应,得到原位生成纳米MnO2胶体,用该胶体颗粒对水中Mn2+进行吸附。分析讨论原位生成的MnO2浓度、吸附温度、吸附时间以及pH值等因素对MnO2对Mn2+离子吸附容量的影响。通过OM、EDS、Zeta电位和FTIR检测分析了MnO2在吸附Mn2+离子前后以及在不同条件下吸附的表面结构和官能团的变化,以期为深入研究MnO2对Mn2+离子的吸附机制提供参考。
Unisense微电极系统的应用
配置浓度为0.1 g/L的Mn2+离子溶液和1.1506 g/L的KMnO4溶液,实验中取9份体积为300 mL浓度为0.1 g/L的Mn2+离子溶液,分别加入对应化学剂量的浓度为1.1506 g/L的KMnO4溶液对Mn2+离子进行氧化吸附,使得原位生成的MnO2胶体浓度分别为1、5、10、20、30、45、60、80、100 mg/L。
使用了Unisense PH微电极以及氧化还原电位电极测试溶液中的PH、还原电位等相关数据,通过分析相应的PH、还原电位等相关数据变化情况,提出了MnO2对Mn2+离子吸附容量的影响机理。
研究结果
Mn2+在25℃下的吸附等温图如图1所示,随着MnO2浓度的升高,其吸附容量逐渐降低,1 mg/L的MnO2溶液中的胶体颗粒吸附容量可达2022.19 mg/g。当MnO2浓度较低时,MnO2颗粒较小,无法团聚形成大颗粒沉淀,仍以胶体的形式存在于Mn2+离子溶液中,其比表面积更大,MnO2表面的活性吸附位点更多,从而更易与Mn2+离子发生吸附反应。而高浓度MnO2条件下,MnO2颗粒之间由于存在竞争吸附,导致其表面吸附位点减少,在吸附位点有限的情况下,这种竞争更加显著;MnO2浓度较低时,溶液中Mn2+离子浓度远高于MnO2表面的浓度,吸附位点与溶液中的Mn2+离子之间的相互作用较强,使得Mn2+离子更容易向MnO2表面扩散,从而增加了吸附速率和吸附容量。
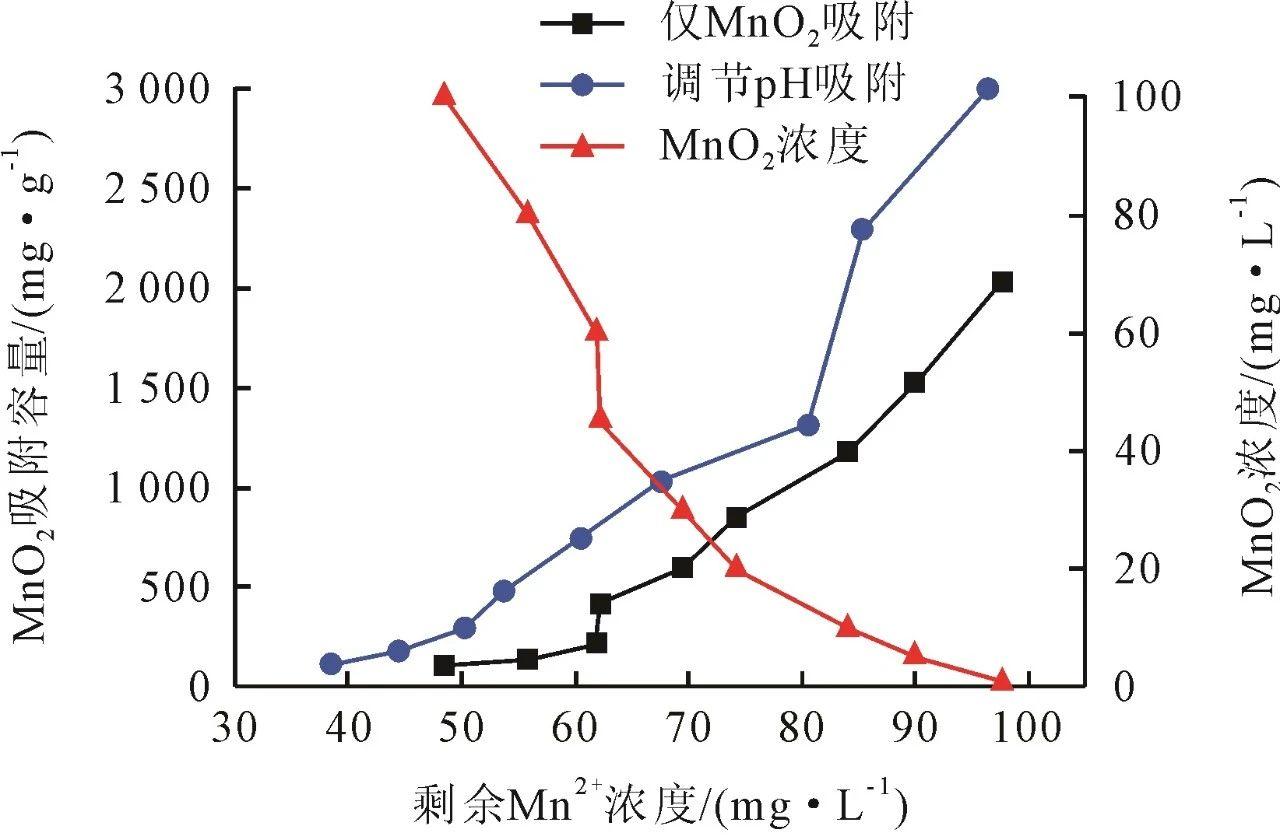
图1原位生成不同浓度的MnO2在25℃下静态吸附和调节pH后的吸附等温图
加入NaOH能显著提高MnO2的吸附容量。分析认为,OH⁻离子能够促使MnO2表面上Mn—OH基团的形成,这些基团能够吸附Mn2+离子,从而提升了MnO2对Mn2+离子的吸附能力。同时OH⁻离子还可以中和MnO2表面的正电荷密度,从而降低了电荷排斥效应,使得MnO2表面更容易吸附带正电的MnO2离子(图2)。
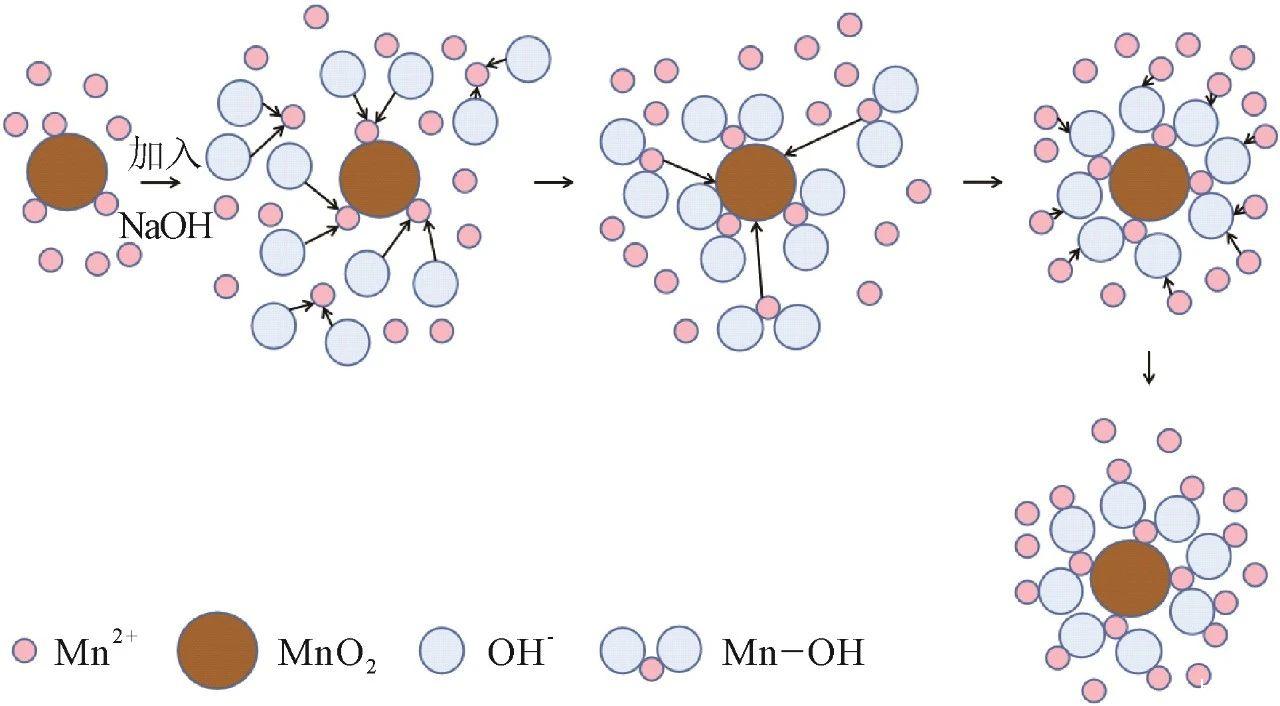
图2调节pH后MnO2对Mn2+离子吸附反应机理
MnO2不同吸附时间对Mn2+离子的吸附容量如图3所示。吸附在30 min左右基本完成,吸附速率由快变慢,30 min后吸附容量有所下降。分析认为,随着吸附的发生,溶液pH会降低,MnO2表面H+离子与吸附的Mn2+竞争吸附位点。当H+离子竞争成功并占据吸附位点时,吸附的Mn2+可能会被释放。综上所述,MnO2吸附Mn2+离子后发生解吸是一个动态平衡过程,受多种因素的影响。这些因素包括吸附位点的饱和程度、竞争吸附离子、溶液的pH等。因此,吸附时间过长会导致Mn2+离子返回溶液中。
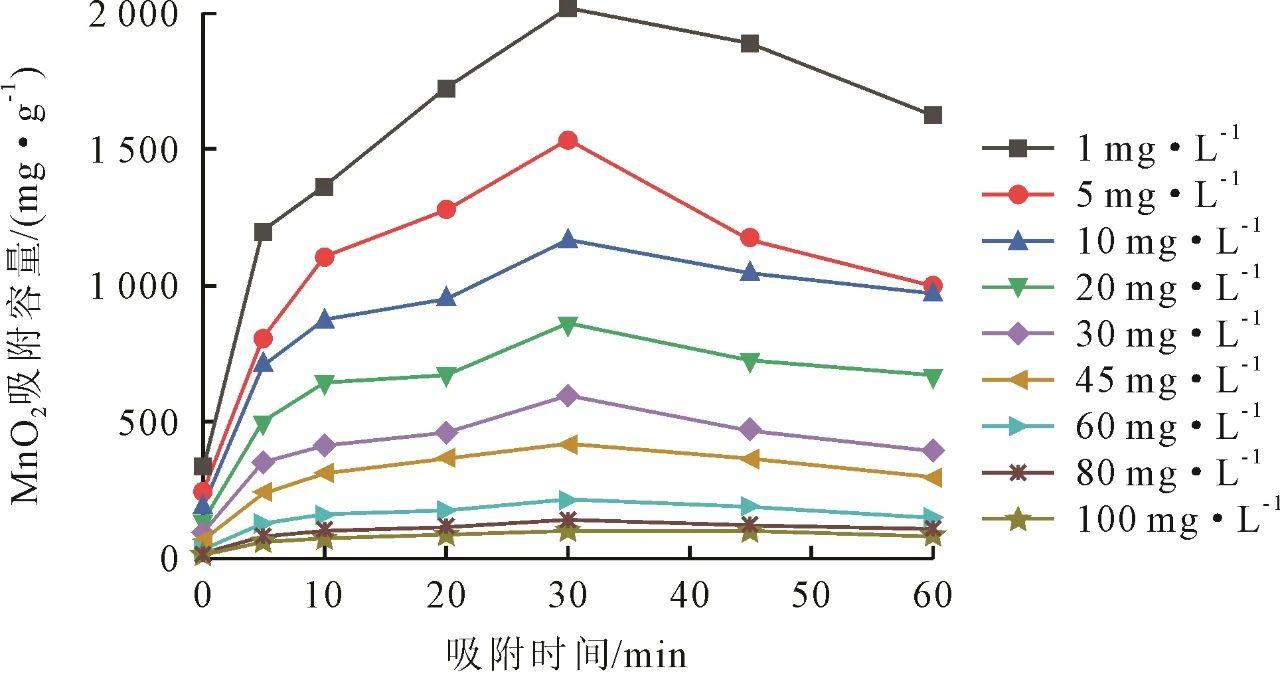
图3 MnO2不同吸附时间对Mn2+离子的吸附容量
MnO2对Mn2+离子的吸附过程符合伪二级动力学方程(图4),MnO2具有多个吸附位点,这些位点上的Mn2+离子能够以不同方式吸附,其表面的吸附位点不仅吸附Mn2+离子,还与其发生化学反应,形成Mn—OH键或其他化学络合物;同时伪二级吸附动力学方程描述了吸附位点饱和的影响,在吸附过程的早期,吸附位点通常是充足的,Mn2+离子可以迅速吸附到MnO2表面。然而,随着吸附的进行,吸附位点可能会逐渐饱和,导致吸附速率减缓,即吸附速率不仅与未被吸附物质浓度有关,还与吸附位点的饱和度有关。
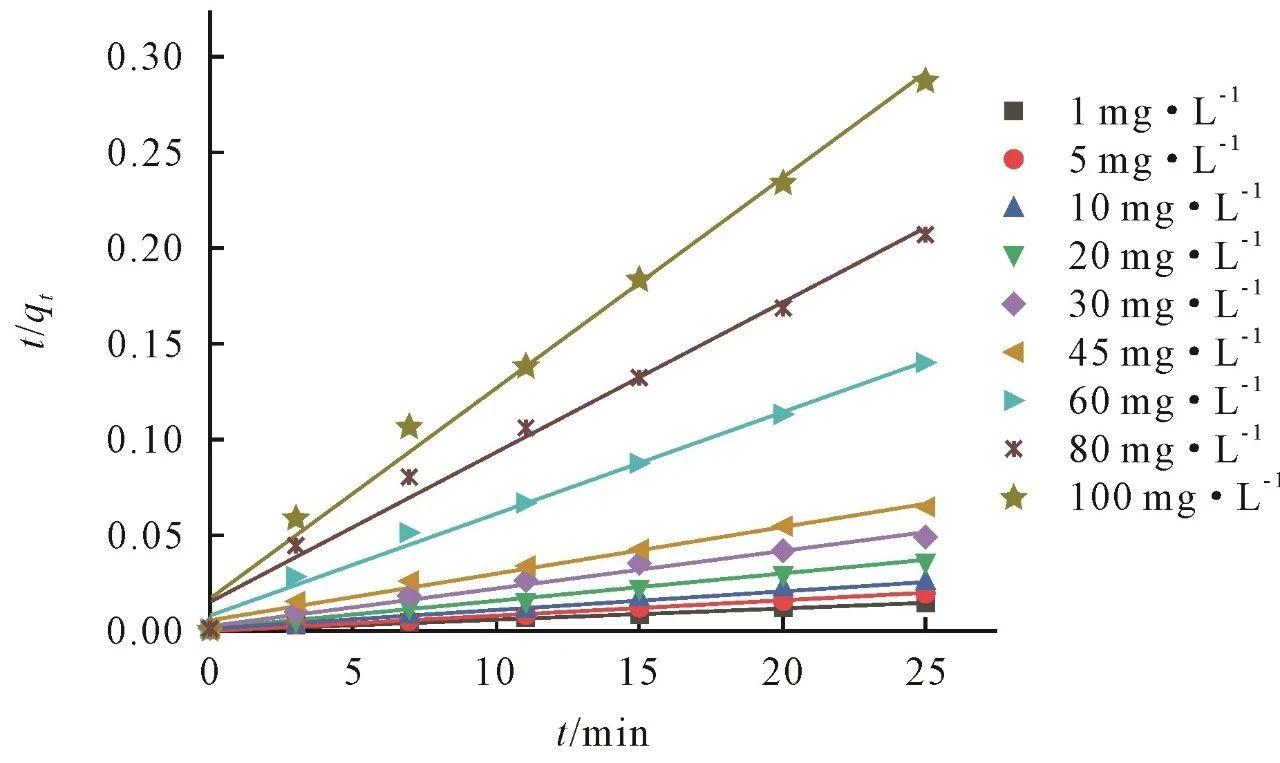
图4伪二级动力学方程拟合
原位生成不同浓度的MnO2分别在5、15、25、35℃下静态吸附等温图如图5所示。低温有利于低浓度MnO2吸附,5℃下浓度为1 mg/L的MnO2吸附容量能达到2638.38 mg/g,但不利于高浓度MnO2吸附。低浓度MnO2比表面积大,吸附位点多,主要依靠物理吸附,物理吸附通常是放热过程,即吸附反应释放热量,故低温下低浓度MnO2吸附容量更大。相反,高浓度的MnO2吸附反应由于温度较低而受到热力学不利因素的影响。
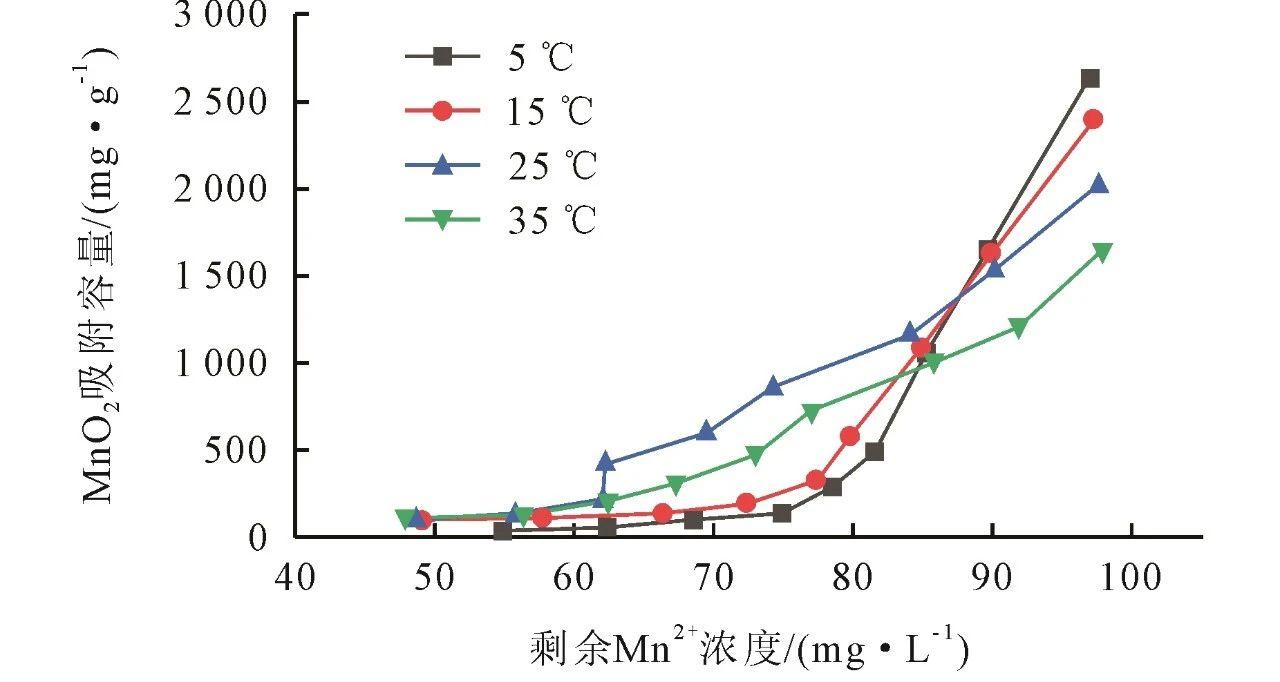
图5原位生成MnO2静态吸附等温图
原位生成的MnO2胶体吸附Mn2+离子前后的形貌图如图6所示。MnO2胶体为准球形,表面粗糙,有大量的孔隙和褶皱。从吸附Mn2+离子后MnO2胶体全貌能够观察到吸附Mn2+离子后的MnO2表面平滑光亮,褶皱基本消失,孔隙大量减少。MnO2表面吸附前后的变化是由于Mn2+的吸附会填充或占据MnO2的孔隙。在吸附过程中,MnO2颗粒的结晶可能会生长,原来的褶皱或不规则结构逐渐被均匀的晶体结构所取代,从而改变了其表面形貌。此外,吸附Mn2+后的MnO2在显微镜下的颜色变成蓝紫色,这是由于吸附Mn2+通常会引起MnO2晶体结构中的电子重新排布,导致过渡态离子的形成,这些过渡态离子吸收400~500 nm波长的光,从而导致蓝紫色的颜色。同时,吸附Mn2+后,MnO2的能带结构可能发生变化,特定的电子跃迁和带隙能量会导致蓝紫色的光吸收和发射。
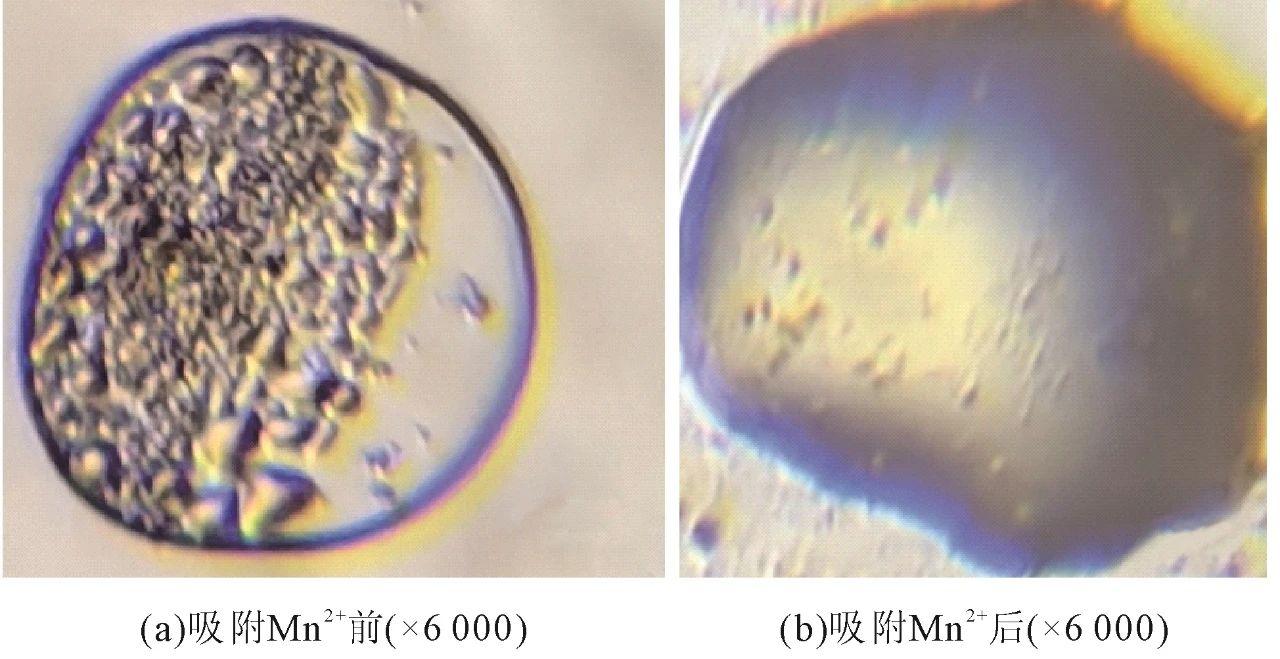
图6原位生成的MnO2在吸附Mn2+离子前后的光学显微镜形貌图
本实验分别测量了吸附前(pH=4.56)以及分别在pH=4.56、pH=7.01、pH=8.03条件下于25℃中吸附后MnO2的Zeta电位,其电势值分别为-30.4、-12.6、-11.7、-1.1 mV。对原位生成的MnO2胶体在pH分别为4.56、7.01、8.03的条件下于25℃下吸附Mn2+离子后进行傅里叶红外光谱检测。随着pH的升高,MnO2表面的羟基振动峰逐渐增强,金属与羟基络合物的相对含量逐渐上升,导致表面Mn—OH含量和表面羟基也相应增多。由此可见,MnO2表面Zeta电位的变化与表面羟基络合有关。
研究结论
原位生成的MnO2胶体上有多个不同类型的吸附位点,每个位点的吸附能力不同,吸附过程不是单一的物理或化学过程,而是一种多层次的吸附现象且吸附可逆。吸附开始时原位生成的MnO2胶体表面的活性位点吸引Mn2+离子,这是吸附过程的初始化阶段。随后,因为MnO2表面活性位点具有负电荷,它们能够吸引带正电的Mn2+离子,Mn2+离子与原位生成的MnO2胶体表面发生电荷交换,并与MnO2表面活性位点上的羟基配位形成Mn—OH吸附在MnO2表面。原位生成纳米MnO2胶体对水中Mn2+离子的吸附实际上是物理吸附、静电吸引、表面配位的共同作用。
